肥厚型心肌病,应该与哪些情况进行鉴别?
心室壁增厚是肥厚型心肌病(HCM)的典型特征,但是心肌肥厚≠肥厚型心肌病。多种生理和病理因素可以导致心室壁增厚,称为HCM的“拟表型”,在临床诊断HCM前需与其他疾病鉴别。
1. 规律锻炼引起的心肌肥厚
长期规律锻炼可以使心脏发生适应性改变,表现为左心室轻度对称性肥厚(通常≤15 mm),但是左心室舒张功能正常,心肺运动功能良好,无心肌病家族史,基因检测阴性。停止锻炼3个月后心肌肥厚程度可以减轻或消退。
2. 高血压引起的心肌肥厚
一般高血压病史较长,长期血压控制不达标,通常为对称性轻度肥厚(≤15 mm),肥厚心肌呈均匀低回声,失代偿期左心室腔可增大。心电图可见左心室高电压,基因检测一般阴性。严格血压控制6~12个月后左心室心肌肥厚可以减轻或消退。
3. 主动脉瓣狭窄引起的心肌肥厚
主动脉瓣狭窄可以引起心脏后负荷增加,导致心肌代偿性肥厚,一般是轻度对称性肥厚(≤15 mm),与HCM存在以下区别:
①收缩期杂音位置较高,以胸骨右缘第2肋间和胸骨左缘第3肋间明显,杂音向颈部传导,改变心脏前后负荷措施对杂音强度影响不大;
②超声心动图检查可见主动脉瓣叶增厚、收缩期开放受限,瓣口面积缩小;
③心导管检查提示左心室与主动脉之间存在收缩期压差,而左心室腔与LVOT之间无压差。
4. 心脏淀粉样变
心脏淀粉样变可主要分为轻链型(AL-CA)和转甲状腺素蛋白型(ATTR-CA)。由于淀粉样物质在心肌细胞外基质沉积,导致心室壁假性肥厚。通常为对称性心室壁增厚,但心电图表现为低电压或正常电压,QRS电压与室壁厚度比值下降;超声心动图可见室间隔和室壁均匀增厚,颗粒状回声增强,房间隔和瓣膜也可以增厚;CMR表现为心内膜下弥漫性甚至透壁性(全层)强化;具有以上警示信号的患者应高度怀疑心脏淀粉样变,需进行血、尿清游离轻链及免疫固定电泳等检查用于AL-CA的筛查和诊断,焦磷酸盐放射性核素骨扫描以及转甲状腺素蛋白基因检测辅助ATTR-CA的诊断和分型;受累器官和/或心内膜活检病理学检查结果为心脏淀粉样变诊断和分型的金标准。
可有心脏外表现:AL型淀粉样变可有50%~70%患者出现肾脏受累,表现为大量蛋白尿,不伴血尿;周围神经受累表现为四肢对称性感觉及运动功能障碍、腕管综合征等,自主神经受累主要表现为位性低血压、排尿困难、排汗异常等;累及消化系统者可出现消化不良综合征及腹泻等临床表现;还可出现巨舌、眶周紫癜等特征性软组织受累表现;此外还可出现肝脏、皮肤、眼部受累等相关症状。
5.先天性代谢性疾病
(1)Fabry病
Fabry病由GLA基因突变引起,遗传模式为X连锁隐性遗传(XR),病因为α-半乳糖苷酶A的基因突变,导致其编码的α-半乳糖苷酶A功能部分或全部缺失,三聚己糖神经酰胺的正常降解受阻,未降解的底物在多种组织的细胞溶酶体中堆积,造成相关组织的功能障碍。α-半乳糖苷酶A酶活性测定及基因检测有助于明确诊断。
心脏受累表现为心肌向心性肥厚,二尖瓣主动脉瓣增厚伴轻中度反流,乳头肌增厚,心电图常表现为左心室高电压及传导系统受累,也可见短PR间期不伴有WPW。CMR检查可见后侧壁中层LGE。
其他系统受累表现如:血管角质瘤、肾脏损害、肢端感觉异常、少汗或无汗、早发脑梗、眩晕或听力障碍、眼膜环形增生及胃肠症状等。
(2)糖原贮积症
常合并多器官受累的临床表现,主要包括以下两类:
Danon病:是一种X连锁显性遗传病,由位于Xq24的LAMP2编码基因突变所致。起病早,男性常于20岁前,女性多于成年期发病。具有典型的三联征表型:心肌肥厚伴遗传性WPW、肌无力和智力发育迟缓。典型病理特征为骨骼肌细胞胞浆内空泡,免疫组化染色提示LAMP-2缺乏。可根据基因检测及特征性病理改变予以鉴别。
Pompe病:婴儿型常累及心肌,多于出生后数月内发病,以心肌肥厚及重度全身性肌张力过低为主要特征,常于1岁前死亡。迟发型者累及心肌者罕见,以骨骼肌病为主,常表现为进行性肌无力。肌肉活检电镜下显示为空泡肌病伴溶酶体内糖原累积,且细胞质中有游离糖原。空泡过碘酸-希夫(PAS)反应阳性,可被淀粉酶消化,且酸性磷酸酶染色阳性。
(3)PRKAG2心脏综合征
PRKAG2心脏综合征由编码单磷酸腺苷激活蛋白激酶γ2亚单位的PRKAG2基因突变引起,遗传模式为常染色体显性遗传。心脏受累表现为心肌肥厚、遗传性WPW、传导系统障碍及室上性心律失常四联征,其他系统表现较少或无。基因检测有助于明确诊断。
6. 神经肌肉疾病
Friedreich共济失调是编码可溶性线粒体蛋白frataxin的FXN基因突变导致,遗传模式为常染色体隐性遗传,心脏受累主要表现为心肌肥厚。基因检测有助于明确诊断。
7. 畸形综合征
包括Noonan综合征(NS)、LEOPARD综合征(LS)等,统称为RAS病。NS和LS由编码蛋白酪氨酸磷酸酶非受体11型的PTPN11基因及原癌基因c-Raf或RAF1及BRAF基因等突变引起,遗传模式为AD。基因检测有助于诊断。
8.线粒体疾病
原发线粒体疾病是由核DNA或线粒体DNA突变所致,常见编码呼吸链蛋白复合物的基因突变,导致能量代谢障碍,出现多系统受累的症状,以对有氧代谢需求高的脑、骨骼肌及心肌表现为主。心脏受累表现以心肌肥厚最常见;其他系统受累表现包括神经肌肉病变、内分泌、消化系统或肾脏等。基因检测有助于确诊。
文献索引:中国医师协会心力衰竭专业委员会, 国家心血管病专家委员会心力衰竭专业委员会, 中华心力衰竭和心肌病杂志编辑委员会. 中国肥厚型心肌病指南2022. 中华心力衰竭和心肌病杂志, 2022, 6(2): 80-103.

- 上一篇
高血压治疗,α受体阻滞剂还有用武之地吗?送你一份用药手册~
α受体阻滞剂是治疗高血压的经典药物,然而目前主要的高血压指南不再推荐其作为一线降压药。α受体阻滞剂在高血压治疗中还有用武之地吗?哪些临床情况下需要使用这类药物呢?近年来,学术界对α受体阻滞剂的临床应用价值有了新的认识,近日国内首部α受体阻滞剂降压治疗共识发布,为临床使用这类药物提供了详细的指导。 共识指出,α受体阻滞剂降压效果肯定,安全性较高,没有明显的代谢
- 下一篇
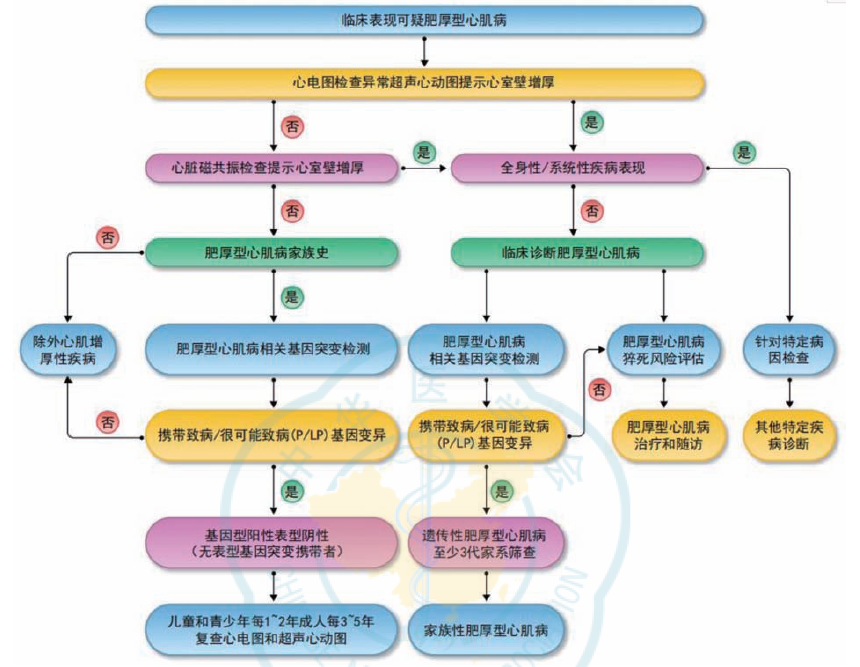
时隔5年,中国肥厚型心肌病管理指南再更新
肥厚型心肌病(HCM)是一种最常见的遗传性心脏病。《中国肥厚型心肌病管理指南2017》发布后的5年来,在HCM的诊断和治疗方面又有了一些新的进展。为此,我国组织HCM相关领域研究的专家对2017版指南进行了更新。《中国肥厚型心肌病管理指南2022》分析总结目前国内外最新研究证据,对HCM的诊断和治疗等内容进行了更新。HCM概述肥厚型心肌病是一种呈常染色体显性